Associate Professor
Nicolaus Copernicus University in Toruń, Institute of Physics, Department of Quantum Physics, Poland

From left to right: Artur Nowak, Aleksandra (Ola) Leszczyk, Katharina (Kasia) Boguslawski, Saddem Mamache, Emil Sujkowski, Maximilian Kriebel, Matheus Moraes. Missing: Marta Gałyńska, Michał Kopczyński
Our current research focuses on geminal-based wavefunction ansätze and alternative Coupled-Cluster models for strongly-correlated systems, the Density Matrix Renormalization Group (DMRG) algorithm, and the application of these methods to challenging problems in chemistry (e.g., chemical bond breaking/forming and actinide chemistry). We also apply intuitive tools to interpret electronic structures and chemical phenomena using concepts of quantum information theory.
Furthermore, together with the research groups of Dariusz Kędziera, Paweł Tecmer, and Piotr Żuchowski, we develop our own open-source quantum chemistry software package called PyBEST, where all our proposed methods are implemented.

From left to right: Artur Nowak, Odile Franck, Aleksandra (Ola) Leszczyk (Łachmańska), Katharina (Kasia) Boguslawski.

I am an associate professor at Nicolaus Copernicus University in Toruń developing new electronic structure method. My current research focuses on geminal-based wavefunction ansätze for strongly-correlated systems and the Density Matrix Renormalization Group (DMRG) algorithm, as well as their application to challenging problems in chemistry (e.g., chemical bond breaking/forming, transition metal complexes and actinide chemistry). In addition to quantum modeling, I am developing intuitive tools to interpret electronic structures and chemical phenomena using concepts from quantum information theory. Moreover, I am a co-founder and lead developer of the PyBEST software package, an open-source, multinational software development project, headquartered at NCU in Torun.
Nicolaus Copernicus University in Toruń, Institute of Physics, Department of Quantum Physics, Poland
Nicolaus Copernicus University in Toruń, Faculty of Chemistry and Institute of Physics, Department of Quantum Physics, Poland
Nicolaus Copernicus University in Toruń, Institute of Physics, Poland
Nicolaus Copernicus University in Toruń, Institute of Physics, Poland.
McMaster University, Department of Chemistry and Chemical Biology, Canada
ETH Zurich, Laboratory of Physical Chemistry, Switzerland
Habilitation in Physics
Institute of Physics, Nicolaus Copernicus University in Toruń, Poland.
Thesis: Modeling strong and weak correlation using electron-pair states
Dr. Sc. ETH Zurich in Chemistry
Laboratory of Physical Chemistry, ETH Zurich, Switzerland
Master of Science in Chemistry
ETH Zurich, Switzerland
Bachelor of Science in Chemistry
ETH Zurich, Switzerland







Master student, Institute of Physics, Nicolaus Copernicus University in Toruń, Poland.
Project title: Dissecting cation-cation interactions using unconventional wavefunction approaches
PhD student, Department of Chemistry and Chemical Biology, McMaster University, Canada.
Project title: DMRG and Tensor Network states in heavy element chemistry
Publication: Theoretical Chemistry Accounts, 134, 120 (2015)
TAPS student from AGH University of Science and Technology, Kraków, Poland.
Project title: Quantum-mechanical modeling of core and core-valence properties of heavy-element compounds
Publication: to be submitted
TAPS student from ETH Zurich.
Project title: Quantum-mechanical modelling of heavy-element compounds: elucidating the activation of the uranyl-oxo bond in gas phase
Publication: to be submitted
Summer Student, Department of Chemistry and Chemical Biology, McMaster University, Canada.
Project title: Thermochemistry of actinide compounds
Publication: Physical Chemistry Chemical Physics, 19, 4317 (2017)
Bachelor Student, Department of Chemistry and Chemical Biology, McMaster University, Canada.
Project title: Cation-cation interactions in actinide oxides
Publication: Physical Chemistry Chemical Physics, 18, 18305-18311 (2016)
Co-op student, Department of Chemistry and Chemical Biology, McMaster University, Canada.
Project title: Electronic structure of iridium complexes
Publication: RSC Advances, 5, 84311–84320 (2015)
Summer Student, Department of Chemistry and Chemical Biology, McMaster University, Canada.
Project title: Metal–olefin bonding in Ni-ethylene complexes
Publication: Chemical Physics Letters 621, 160-164 (2015)
Semester Student, Laboratory of Physical Chemistry, ETH Zurich, Switzerland.
Project title: Bond breaking processes of Carbon/Carbon and Carbon/Homologues — A quantum entanglement study
Publication: Physical Chemistry Chemical Physics 16 (19), 8872-8880 (2014)
Semester Student, Laboratory of Physical Chemistry, ETH Zurich, Switzerland.
Project title: Assessment of extrapolation schemes for the DMRG algorithm
Introduction to CI, MCSCF, PT, and CC theory and their applications.
Introduction to HF and DFT and their applications.
Introduction to DFT, CC, and MCSCF and their applications.
Introduction to DFT, CC, and MCSCF and their applications.
Introduction to DFT, CC, and MCSCF and their applications.
Kinetics, Surface- and Transport-phenomena.
Introduction to Computer Science.
Physical Chemistry.
Introduction to Quantum Chemistry.
Relativistic Quantum Chemistry.
Topic: Spin-orbit coupling
Topic: Development of dressed-pCCD-based methods
Topic: Geometry optimization and property calculations with pCCD
Topic: Spin-flip-type methods with pCCD
Topic: dressed-pCCD
Topic: DEA-pCCD-based methods
Topic: GPU acceleration
Topic: Testing framework within PyBEST
• Marta Gałyńska (NAWA Ulam/Seal of Excellence, October 2022-September 2024)
• Maximilian Hubertus Kriebel (SGS call 2020/NCBR, March 2022-March 2024)
• Odile Franck (NCN SONATA BIS, January 2017-March 2018)
• Mostafa Javaheri Moghadam (visiting from New Brunswick University, Canada, April 2023-July 2023)
• Matheus Moraes (visiting from Federal University of ABC, Brazil, May 2022-October 2022)
• Saddem Mamache (PhD candidate, April 2022-March 2024)
• Aleksandra (Ola) Leszczyk (October 2017-November 2022)
• Artur Nowak (March 2017-September 2022)
• Igor Sawicki (Bachelor/Master student, April 2023-June 2024)
• Michał Suchorowski (TAPS student, August/September 2019)
• Tibor Dome (TAPS student, July 2018)
Our research focuses on the development of new quantum chemistry methods based on electron-pair states for ground and excited states of closed- and open-shell compounds, computational studies on molecular properties and chemical reactivity using tensor-network states approaches, and the design of interpretive tools for a qualitative understanding of chemical compounds and chemical processes using quantum information theoretical concepts.
The head of the group is also a developer of the Horton program package, which supports Hartree-Fock, DFT, and post-Hartree-Fock calculations (MP2, AP1roG, PTa, PTb, LCC) and features various wavefunction post-processing tools.
A detailed description of our research interests is summarized below.

We are developing new methods for strong and weak electron correlation based on electron-pair states, called geminals. In geminal-based methods, the electronic wavefunction is constructed as an antisymmetric product of two-electron functions. By construction, this captures the essential part of strong correlation. I presented the first implementation of the Antisymmetric Product of 1-reference orbital Geminals (AP1roG) for molecules including a fully variational optimization scheme for the molecular orbitals using a Lagrangian function. We further developed different flavours of non-variational orbital optimization techniques based on the seniority concept. Although geminal-based methods are able to describe strong electron correlation, they miss a large fraction of the weak electron correlation energy. Recently, we presented a multi-reference linearized coupled-cluster correction based on an AP1roG reference function to include weak correlation on top of an AP1roG wavefunction. Specifically, the linearized coupled-cluster model outperformed various perturbation theory corrections previously developed for AP1roG.
Selected key publications are:

We are extending the geminal-based wavefunction models we are developing to excited states. Specifically, we demonstrated that AP1roG-based methods can reduce statistical errors by a factor of 2 compared to conventional coupled cluster methods of similar cost.
Selected key publications are:
Together with our collaborators, we are applying our geminal-based wavefunction models to weakly interacting systems. Specifically, we demonstrated that AP1roG-based methods can reduce statistical errors in interaction energies compared to the conventional CCSD ansatz.
Selected key publications are:

Tensor Network States approaches, like the DMRG algorithm, are a promising alternative to standard electron correlation methods. We are investigating the performance of DMRG to accurately predict electronic structures and molecular properties. Specifically, we developed a reconstruction algorithm to construct configuration-interaction-type wavefunctions from the matrix-product-state representation optimized by DMRG using a Monte-Carlo sampling algorithm. Furthermore, we implemented and studied the calculation of spin density and magnetization density distributions from DMRG wavefunctions.
Selected key publications are:

The interaction of orbitals is a useful concept in chemistry. It is frequently used to understand chemical processes and reaction mechanisms. Unfortunately, the interaction of orbitals is commonly understood using qualitative arguments, like molecular-orbital diagrams, Frontier-orbital theory, and ligand field theory. We are developing quantitative means to measure the interaction of orbitals using concepts of quantum information theory. Specifically, we have shown that orbital entanglement and correlation are particularly useful and intuitive measures to quantify the interaction of orbitals and to elucidate electronic structures and changes in electronic structure that accompany chemical processes. Furthermore, we presented various applications where orbital entanglement was used to identify bond orders, transition states, and electron correlation effects.
Selected key publications are:

Our research also focuses on theoretical modeling of heavy-element compounds and their properties using conventional and unconventional electron correlation methods. Specifically, we presented the first DMRG study on actinide chemistry, where we investigated the anticipated singlet-triplet spin crossover of the CUO molecule diluted in a noble gas matrix (within a scalar relativistic treatment) and elucidated the mysterious interaction of the CUO unit with the noble gas environment. Most importantly, this was the first theoretical study confirming the experimentally anticipated singlet-triplet ground state change of the CUO molecule. Furthermore, we presented the first geminal study on actinide chemistry. Specifically, AP1roG was the first quantum chemistry method to completely dissociate the UO22+ molecule into its atomic fragments.
Selected key publications are:

We are actively involved in software development. The head of the group is one of the founders and lead developers of the PyBEST program package, an open-source quantum chemistry software package written in Python and C++. Specifically, she is the main developer of the post-Hartree-Fock modules of the PyBEST program package, which includes
You can find more information and download PyBEST here Or download various versions from zenodo or PyPI.
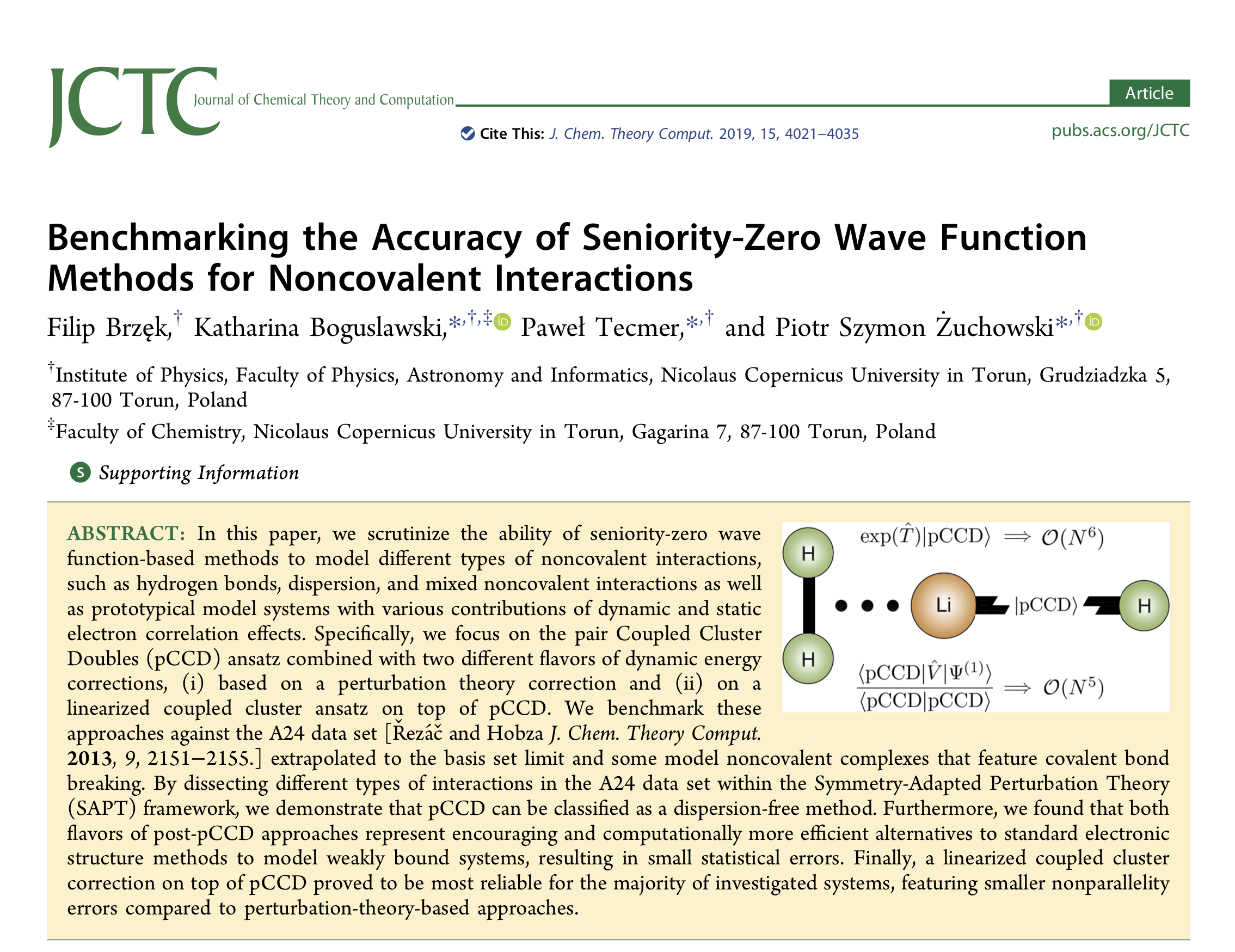
In this paper, we scrutinize the ability of seniority-zero wave function-based methods to model different types of noncovalent interactions, such as hydrogen bonds, dispersion, and mixed noncovalent interactions as well as prototypical model systems with various contributions of dynamic and static electron correlation effects. Specifically, we focus on the pair Coupled Cluster Doubles (pCCD) ansatz combined with two different flavors of dynamic energy corrections, (i) based on a perturbation theory correction and (ii) on a linearized coupled cluster ansatz on top of pCCD. We benchmark these approaches against the A24 data set [Řezáč and Hobza J. Chem. Theory Comput. 2013, 9, 2151−2155.] extrapolated to the basis set limit and some model noncovalent complexes that feature covalent bond breaking. By dissecting different types of interactions in the A24 data set within the Symmetry-Adapted Perturbation Theory (SAPT) framework, we demonstrate that pCCD can be classified as a dispersion-free method. Furthermore, we found that both flavors of post-pCCD approaches represent encouraging and computationally more efficient alternatives to standard electronic structure methods to model weakly bound systems, resulting in small statistical errors. Finally, a linearized coupled cluster correction on top of pCCD proved to be most reliable for the majority of investigated systems, featuring smaller nonparallelity errors compared to perturbation-theory-based approaches.
The accurate description of doubly excited states using conventional electronic structure methods is remarkably challenging, primarily because such excited states require the inclusion of doubly or higher excited configurations or the application of multireference methods. We present a new approach to target electronically excited states that feature a double-electron transfer. Our method uses the equation of motion (EOM) formalism with a pair coupled cluster doubles (pCCD) reference function, where dynamical correlation is accounted for by a linearized coupled cluster correction with singles and doubles (LCCSD). Specifically, our proposed EOM-pCCD-LCCSD model represents a simplification of the conventional EOM-CCSD approach, where the electron-pair amplitudes of CCSD are tailored by pCCD. The performance of EOM-pCCD-LCCSD is assessed for the lowest-lying excited states in CH+ and all-trans polyenes. In contrast to conventional EOM-CC methods with at most double excitations, EOM-pCCD-LCCSD predicts the right order of states in polyenes with excitation energies closest to experiment, outperforming even highly accurate methods such as the density matrix renormalization group algorithm.
Understanding the binding mechanism in neptunyl clusters formed due to cation–cation interactions is of crucial importance in nuclear waste reprocessing and related areas of research. Since experimental manipulations with such species are often rather limited, we have to rely on quantum-chemical predictions of their electronic structures and spectroscopic parameters. In this work, we present a state-of-the-art quantum chemical study of the T-shaped and diamond-shaped neptunyl(V) and neptunyl(VI) dimers. Specifically, we scrutinize their molecular structures, (implicit and explicit) solvation effects, the interplay of static and dynamical correlation, and the influence of spin–orbit coupling on the ground state and lowest-lying excited states for different total spin states and total charges of the neptunyl dications. Furthermore, we use the picture of interacting orbitals (quantum entanglement and correlation analysis) to identify strongly correlated orbitals in the cation–cation complexes that should be included in complete active space calculations. Most importantly, our study highlights the complex interplay of correlation effects and relativistic corrections in the description of the ground and lowest-lying excited states of neptunyl dications.
Wave functions restricted to electron-pair states are promising models to describe static/nondynamic electron correlation effects encountered, for instance, in bond-dissociation processes and transition-metal and actinide chemistry. To reach spectroscopic accuracy, however, the missing dynamic electron correlation effects that cannot be described by electron-pair states need to be included a posteriori. In this Article, we extend the previously presented perturbation theory models with an Antisymmetric Product of 1-reference orbital Geminal (AP1roG) reference function that allows us to describe both static/nondynamic and dynamic electron correlation effects. Specifically, our perturbation theory models combine a diagonal and off-diagonal zero-order Hamiltonian, a single-reference and multireference dual state, and different excitation operators used to construct the projection manifold. We benchmark all proposed models as well as an a posteriori Linearized Coupled Cluster correction on top of AP1roG against CR-CC(2,3) reference data for reaction energies of several closed-shell molecules that are extrapolated to the basis set limit. Moreover, we test the performance of our new methods for multiple bond breaking processes in the homonuclear N2, C2, and F2 dimers as well as the heteronuclear BN, CO, and CN+ dimers against MRCI-SD, MRCI-SD+Q, and CR-CC(2,3) reference data. Our numerical results indicate that the best performance is obtained from a Linearized Coupled Cluster correction as well as second-order perturbation theory corrections employing a diagonal and off-diagonal zero-order Hamiltonian and a single-determinant dual state. These dynamic corrections on top of AP1roG provide substantial improvements for binding energies and spectroscopic properties obtained with the AP1roG approach, while allowing us to approach chemical accuracy for reaction energies involving closed-shell species.
Wavefunctions restricted to electron pair states are promising models for strongly-correlated systems. Specifically, the pair Coupled Cluster Doubles (pCCD) ansatz allows us to accurately describe bond dissociation processes and heavy-element containing compounds with multiple quasi-degenerate single-particle states. Here, we extend the pCCD method to model excited states using the equation of motion (EOM) formalism. As the cluster operator of pCCD is restricted to electron-pair excitations, EOM-pCCD allows us to target excited electron-pair states only. To model singly excited states within EOM-pCCD, we modify the configuration interaction ansatz of EOM-pCCD to contain also single excitations. Our proposed model represents a simple and cost-effective alternative to conventional EOM-CC methods to study singly excited electronic states. The performance of the excited state models is assessed against the lowest-lying excited states of the uranyl cation and the two lowest-lying excited states of all-trans polyenes. Our numerical results suggest that EOM-pCCD including single excitations is a good starting point to target singly excited states.
Wave functions constructed from electron-pair states can accurately model strong electron correlation effects and are promising approaches especially for larger many-body systems. In this article, we analyze the nature and the type of electron correlation effects that can be captured by wave functions restricted to electron-pair states. We focus on the pair-coupled-cluster doubles (pCCD) ansatz also called the antisymmetric product of the 1-reference orbital geminal (AP1roG) method, combined with an orbital optimization protocol presented in Boguslawski et al. [Phys. Rev. B 89, 201106(R) (2014)], whose performance is assessed against electronic structures obtained form density-matrix renormalization-group reference data. Our numerical analysis covers model systems for strong correlation: the one-dimensional Hubbard model with a periodic boundary condition as well as metallic and molecular hydrogen rings. Specifically, the accuracy of pCCD/AP1roG is benchmarked using the single-orbital entropy, the orbital-pair mutual information, as well as the eigenvalue spectrum of the one-orbital and two-orbital reduced density matrices. Our study indicates that contributions from singly occupied states become important in the strong correlation regime which highlights the limitations of the pCCD/AP1roG method. Furthermore, we examine the effect of orbital rotations within the pCCD/AP1roG model on correlations between orbital pairs.
We present a Linearized Coupled Cluster (LCC) correction based on an Antisymmetric Product of 1-reference orbital Geminals (AP1roG) reference state. In our LCC ansatz, the cluster operator is restricted to double or to single and double excitations, as in standard single-reference CC theory. The performance of the AP1roG-LCC models is tested for the dissociation of diatomic molecules in their lowest-lying singlet state (C2, F2, and BN), the symmetric dissociation of the H50 hydrogen chain, and spectroscopic constants of the uranyl cation (UO22+). Our study indicates that an LCC correction based on an AP1roG reference function is more robust and reliable than corrections based on perturbation theory, yielding spectroscopic constants that are in very good agreement with theoretical reference data.
The basic concepts of orbital entanglement and its application to chemistry are briefly reviewed. The calculation of orbital entanglement measures from correlated wavefunctions is discussed in terms of reduced n-particle density matrices. Possible simplifications in their evaluation are highlighted in case of seniority-zero wavefunctions. Specifically, orbital entanglement allows us to dissect electron correlation effects in its strong and weak contributions, to determine bond orders, to assess the quality and stability of active space calculations, to monitor chemical reactions, and to identify points along the reaction coordinate where electronic wavefunctions change drastically. Thus, orbital entanglement represents a useful and intuitive tool to interpret complex electronic wavefunctions and to facilitate a qualitative understanding of electronic structure and how it changes in chemical processes.
The efficient and accurate description of the electronic structure of strongly correlated systems is still a largely unsolved problem. The usual procedures start with a multiconfigurational (usually a Complete Active Space, CAS) wavefunction which accounts for static correlation and add dynamical correlation by perturbation theory, configuration interaction, or coupled cluster expansion. This procedure requires the correct selection of the active space. Intuitive methods are unreliable for complex systems. The inexpensive black-box unrestricted natural orbital (UNO) criterion postulates that the Unrestricted Hartree-Fock (UHF) charge natural orbitals with fractional occupancy (e.g., between 0.02 and 1.98) constitute the active space. UNOs generally approximate the CAS orbitals so well that the orbital optimization in CAS Self-Consistent Field (CASSCF) may be omitted, resulting in the inexpensive UNO-CAS method. A rigorous testing of the UNO criterion requires comparison with approximate full configuration interaction wavefunctions. This became feasible with the advent of Density Matrix Renormalization Group (DMRG) methods which can approximate highly correlated wavefunctions at affordable cost. We have compared active orbital occupancies in UNO-CAS and CASSCF calculations with DMRG in a number of strongly correlated molecules: compounds of electronegative atoms (F2, ozone, and NO2), polyenes, aromatic molecules (naphthalene, azulene, anthracene, and nitrobenzene), radicals (phenoxy and benzyl), diradicals (o-, m-, and p-benzyne), and transition metal compounds (nickel-acetylene and Cr2). The UNO criterion works well in these cases. Other symmetry breaking solutions, with the possible exception of spatial symmetry, do not appear to be essential to generate the correct active space. In the case of multiple UHF solutions, the natural orbitals of the average UHF density should be used. The problems of the UNO criterion and their potential solutions are discussed: finding the UHF solutions, discontinuities on potential energy surfaces, and inclusion of dynamical electron correlation and generalization to excited states.
We introduce new nonvariational orbital optimization schemes for the antisymmetric product of one-reference orbital geminal (AP1roG) wave function (also known as pair-coupled cluster doubles) that are extensions to our recently proposed projected seniority-two (PS2-AP1roG) orbital optimization method [J. Chem. Phys. 2014, 140, 214114)]. These approaches represent less stringent approximations to the PS2-AP1roG ansatz and prove to be more robust approximations to the variational orbital optimization scheme than PS2-AP1roG. The performance of the proposed orbital optimization techniques is illustrated for a number of well-known multireference problems: the insertion of Be into H2, the automerization process of cyclobutadiene, the stability of the monocyclic form of pyridyne, and the aromatic stability of benzene.
We present a new, non-variational orbital-optimization scheme for the antisymmetric product of one-reference orbital geminal wave function. Our approach is motivated by the observation that an orbital-optimized seniority-zero configuration interaction (CI) expansion yields similar results to an orbital-optimized seniority-zero-plus-two CI expansion [L. Bytautas, T. M. Henderson, C. A. Jimenez-Hoyos, J. K. Ellis, and G. E. Scuseria, J. Chem. Phys.135, 044119 (2011)]. A numerical analysis is performed for the C2 and LiF molecules, for the CH2 singlet diradical as well as for the symmetric stretching of hypothetical (linear) hydrogen chains. For these test cases, the proposed orbital-optimization protocol yields similar results to its variational orbital optimization counterpart, but prevents symmetry-breaking of molecular orbitals in most cases.
 This chapter discusses chemical bonding in open-shell molecules that can be attributed to its unpaired electrons. We first give a short overview of contemporary single- and multireference quantum chemical methods that can be employed for quantitative bond energy calculations. Quantum chemistry provides us with electronic wave function and (spin) density, which represent the central ingredients for a subsequent analysis of the chemical bond. In this respect, we review different strategies that have been developed for a qualitative interpretation of open-shell electronic structures. A key feature of such approaches is the introduction of local quantities such as local spins. Different decomposition schemes of the total spin expectation value for multireference wave functions are presented and compared. In particular, the localization of spins facilitates the description of magnetically coupled centers. Such coupling phenomena generally require a multideterminant treatment, yet a one-determinant picture can be enforced if the spin symmetry of the system is broken. Different approaches that optimize such broken-symmetry solutions are discussed. Furthermore, various definitions of the covalent bond order applicable to open-shell electronic structures are discussed, which are based either on the (spin) density matrix or on a simple electron-counting scheme. While the former suffers from its method and basis-set dependence, the latter represents a reliable estimate only if one considers all bonding and antibonding orbitals that are crucial for a correct description of the chemical bond.
This chapter discusses chemical bonding in open-shell molecules that can be attributed to its unpaired electrons. We first give a short overview of contemporary single- and multireference quantum chemical methods that can be employed for quantitative bond energy calculations. Quantum chemistry provides us with electronic wave function and (spin) density, which represent the central ingredients for a subsequent analysis of the chemical bond. In this respect, we review different strategies that have been developed for a qualitative interpretation of open-shell electronic structures. A key feature of such approaches is the introduction of local quantities such as local spins. Different decomposition schemes of the total spin expectation value for multireference wave functions are presented and compared. In particular, the localization of spins facilitates the description of magnetically coupled centers. Such coupling phenomena generally require a multideterminant treatment, yet a one-determinant picture can be enforced if the spin symmetry of the system is broken. Different approaches that optimize such broken-symmetry solutions are discussed. Furthermore, various definitions of the covalent bond order applicable to open-shell electronic structures are discussed, which are based either on the (spin) density matrix or on a simple electron-counting scheme. While the former suffers from its method and basis-set dependence, the latter represents a reliable estimate only if one considers all bonding and antibonding orbitals that are crucial for a correct description of the chemical bond.
We present an efficient approach to the electron correlation problem that is well suited for strongly interacting many-body systems, but requires only mean-field-like computational cost. The performance of our approach is illustrated for one-dimensional Hubbard rings with different numbers of sites, and for the nonrelativistic quantum-chemical Hamiltonian exploring the symmetric dissociation of the H50 hydrogen chain.
We present a systematic theoretical study on the dissociation of diatomic molecules and their spectroscopic constants using our recently presented geminal-based wave function ansätze. Specifically, the performance of the antisymmetric product of rank two geminals (APr2G), the antisymmetric product of 1-reference-orbital geminals (AP1roG) and its orbital-optimized variant (OO-AP1roG) are assessed against standard quantum chemistry methods. Our study indicates that these new geminal-based approaches provide a cheap, robust, and accurate alternative for the description of bond-breaking processes in closed-shell systems requiring only mean-field-like computational cost. In particular, the spectroscopic constants obtained from OO-AP1roG are in very good agreement with reference theoretical and experimental data.
The chemical bond is an important local concept to understand chemical compounds and processes. Unfortunately, like most local concepts, the chemical bond and the bond order do not correspond to any physical observable and thus cannot be determined as an expectation value of a quantum chemical operator. We recently demonstrated [Boguslawski et al., J. Chem. Theory Comput., 2013, 9, 2959–2973] that one- and two-orbital-based entanglement measures can be applied to interpret electronic wave functions in terms of orbital correlation. Orbital entanglement emerged as a powerful tool to provide a qualitative understanding of bond-forming and bond-breaking processes, and allowed for an estimation of bond orders of simple diatomic molecules beyond the classical bonding models. In this article we demonstrate that the orbital entanglement analysis can be extended to polyatomic molecules to understand chemical bonding.
The accurate calculation of the (differential) correlation energy is central to the quantum chemical description of bond-formation and bond-dissociation processes. In order to estimate the quality of single- and multireference approaches for this purpose, various diagnostic tools have been developed. In this work, we elaborate on our previous observation [J. Phys. Chem. Lett.2012, 3, 3129] that one- and two-orbital-based entanglement measures provide quantitative means for the assessment and classification of electron correlation effects among molecular orbitals. The dissociation behavior of some prototypical diatomic molecules features all types of correlation effects relevant for chemical bonding. We demonstrate that our entanglement analysis is convenient to dissect these electron correlation effects and to provide a conceptual understanding of bond-forming and bond-breaking processes from the point of view of quantum information theory.
Electron correlation effects are essential for an accurate ab initio description of molecules. A quantitative a priori knowledge of the single- or multireference nature of electronic structures as well as of the dominant contributions to the correlation energy can facilitate the decision regarding the optimum quantum chemical method of choice. We propose concepts from quantum information theory as orbital entanglement measures that allow us to evaluate the single- and multireference character of any molecular structure in a given orbital basis set. By studying these measures we can detect possible artifacts of small active spaces.
We present a procedure to construct a configuration-interaction expansion containing arbitrary excitations from an underlying full-configuration-interaction-type wave function defined for a very large active space. Our procedure is based on the density-matrix renormalization group (DMRG) algorithm that provides the necessary information in terms of the eigenstates of the reduced density matrices to calculate the coefficient of any basis state in the many-particle Hilbert space. Since the dimension of the Hilbert space scales binomially with the size of the active space, a sophisticated Monte Carlo sampling routine is employed. This sampling algorithm can also construct such configuration-interaction-type wave functions from any other type of tensor network states. The configuration-interaction information obtained serves several purposes. It yields a qualitatively correct description of the molecule’s electronic structure, it allows us to analyze DMRG wave functions converged for the same molecular system but with different parameter sets (e.g., different numbers of active-system (block) states), and it can be considered a balanced reference for the application of a subsequent standard multi-reference configuration-interaction method.
A refined implicit aqueous solvation model is proposed for the simulation of biomolecules without the explicit inclusion of the solvent degrees of freedom. The mean force due to solvation is approximated by the derivative of a simple analytic function of the solvent accessible surface area combined with two atomic solvation parameters, as described previously, with the addition of a novel term to account for the interaction of the interior atoms of the solute with the solvent. The extended model is parametrized by comparing the structural properties and energies computed from simulations of six test proteins of varying sizes and shapes using the new solvation energy term with the corresponding values obtained from simulations in vacuum, using the original implicit solvent model and in explicit water, and from the X-ray or NMR model structures. The mean solvation model proposed here improves the structural properties relative to vacuum simulations and relative to the simpler model that neglects the volume contribution, while remaining significantly more efficient than simulations in explicit water.
• Anna Krylov, University of Southern California, USA
• Valerie Vallet, CNRS, Universite Lille, Lille, France
• Andre Severo Pereira Gomes, CNRS, Universite Lille, Lille, France
• Florent Real, CNRS, Universite Lille, Lille, France
• Örs Legeza, Wigner Research Center for Physics, Budapest, Hungary
• Gergely Barcza, Wigner Research Center for Physics, Budapest, Hungary
• Stijn De Beardemacker, University of New Brunswick, Canada
• Dimitri Van Neck, Center for Molecular Modeling, Ghent University, Ghent, Belgium
• Patrick Bultinck, Center for Molecular Modeling, Ghent University, Ghent, Belgium
• Toon Verstraelen, Center for Molecular Modeling, Ghent University, Ghent, Belgium
We are looking for highly motivated PhD students and postdoctoral fellows.
We also welcome all Bachelor and Master students who are interested in working on a research project in my group.
See below for more informations.
Currently, we do not offer funding for postdoctoral fellows. However, there are numerous posibilities to apply for external funding.
If you are interested to join my group and
send me a CV and a description of your own research interests.
We also welcome applications from postdoctoral candidates with a background in theoretical chemistry and computational science who are interested in many-body electronic structure theory and the application of computer science tools to challenging electronic structure problems.
If you are interested in
then please email me. I will ensure that you will learn about
Are you intrigued by software development in Python?
Are you curious about many-body electronic structure theory?
Would you like to learn more about conventional and unconventional electronic structure methods?
Are you interested in applying existing quantum-chemistry programs to challening problems in heavy-element chemistry, including the description of relativistic effects and the correlated motion of electrons?
If so, you are welcome to join our group. Please, contact me if you consider working on a research project or your Master thesis.
If you have any questions, feel free to come by my office or contact me.
You can find me at my office located at the Department of Quantum Physics, Institute of Physics, Nicolaus Copernicus University in Toruń, room 480.
If I am not around, please, write me an email to fix an appointment.
Katharina Boguslawski
Department of Quantum Physics
Institute of Physics
Faculty of Physics, Astronomy and Informatics
Nicolaus Copernicus University in Torun
ul. Grudziadzka 5/7
87-100 Torun
Poland
![]()

![]()


